



CAUSAS DE LA ACONDROPLASIA
- Inicio
- MEDICINA Y CIENCIA
- CAUSAS DE LA ACONDROPLASIA
ORIGEN DE LA ACONDROPLASIA

ORÍGEN GENÉTICO DEL ENANISMO
La acondroplasia está originada por una modificación genética autosómica dominante en todas las células del embrión que se manifiesta principalmente como una disfunción del desarrollo del esqueleto (displasia ósea). Esta displasia está originada por una diferenciación acelerada de las maduración de los condrocitos y por tanto una conversión acelerada de cartílago en hueso.
La mutación aparece en alguna de las células progenitoras de forma espontánea (sin que haya mutaciones en los padres) en el 80% de los casos. La mutación se localiza en el cromosoma 4, en el locus (4p16.3) y consiste en el cambio de la Guanina (G) en posición 1138 por una Adenina (A) o por una Cisteína (C) en el 98 % de los casos que se han secuenciado hasta la fecha.
La transcripción del código genético (ADN) del locus 4p16.3 en el mensaje (ARN) y posterior traducción del mensaje, origina una proteína llamada Receptor del factor de fibroblastos 3 (FGFR3). La mutación en el código genético originará una proteína FGFR3 mutante (que tiene un cambio en el aminoácido 380 de su secuencia, glicina (Gly ó G) por arginina (Arg ó R) (Gly380Arg) ó (G380R) (Figura 2).
El FGFR3 es una proteína transmembranar celular de tipo receptor tirosina quinasa que se expresa en todos los tejidos del organismo durante la vida del individuo.
El FGFR3 se expresa en las membranas de los condrocitos y desempeña un papel importante en su función y por tanto en el desarrollo de los huesos largos (He L et al., 2010).
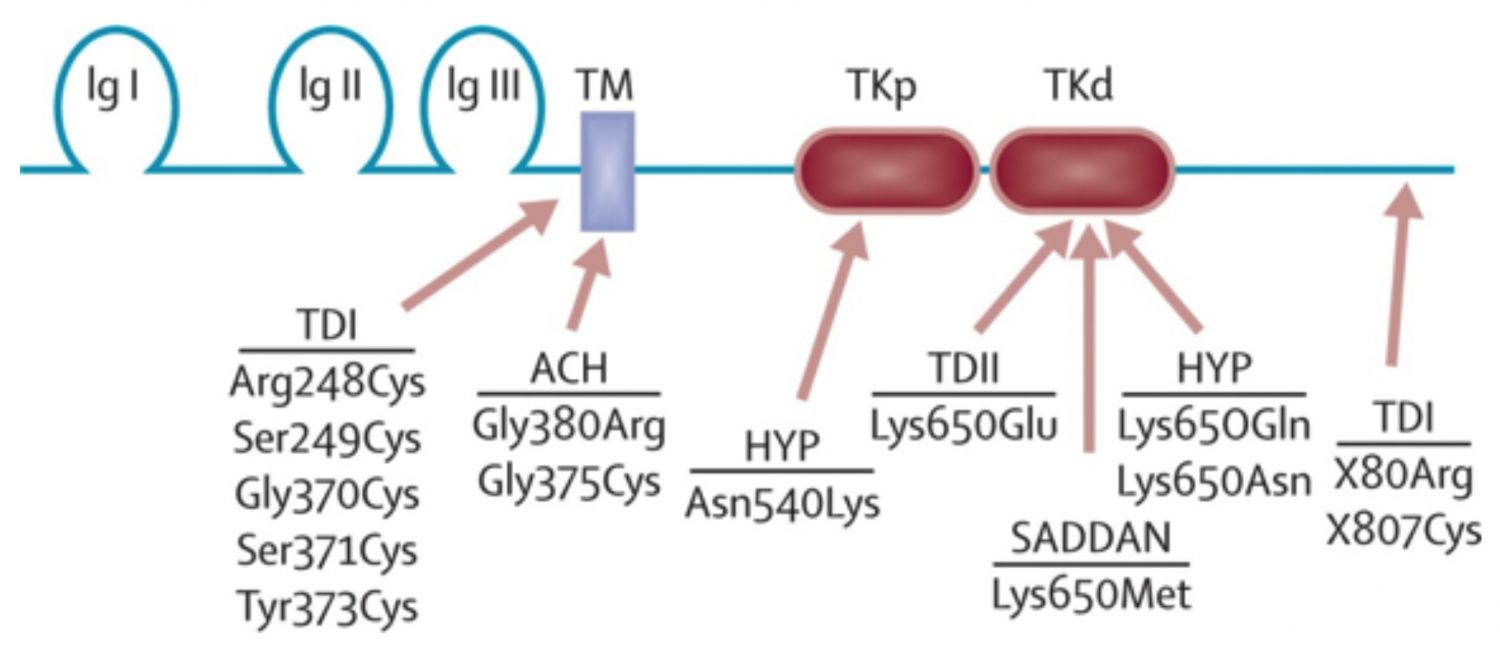
Imagen 2. . Estructura de la proteína FGFR3 que muestra las principales mutaciones en las displasias óseas. ACH = acondroplasia. HYP = hipocondroplasia, TD = displasia tanatafórica. SADDAN = acondroplasia severa con retraso en el desarrollo y acantosis nigricans. Ig = Bucle de inmunoglobulina. TKp / d = dominios de tirosina quinasa proximal y distal. TM = transmembrana. Créditos: Horton W et al., 2007
El FGFR3 transmite señales que regulan la proliferación, maduración y supervivencia de los condrocitos, las células responsables del crecimiento óseo, que se encuentran en las placas de crecimiento de los huesos en crecimiento.
Las señales transmitidas por el FGFR3 se denominan señalización de proteína quinasa activada por mitógeno (MAPK) (Figura 3).
El FGFR3 mutante en acondroplasia tiene una actividad aumentada de señalización MAPK , lo que origina parada en la proliferación de los condrocitos y diferenciación de los mismos en células óseas (Narayana J y Horton W, 2013).
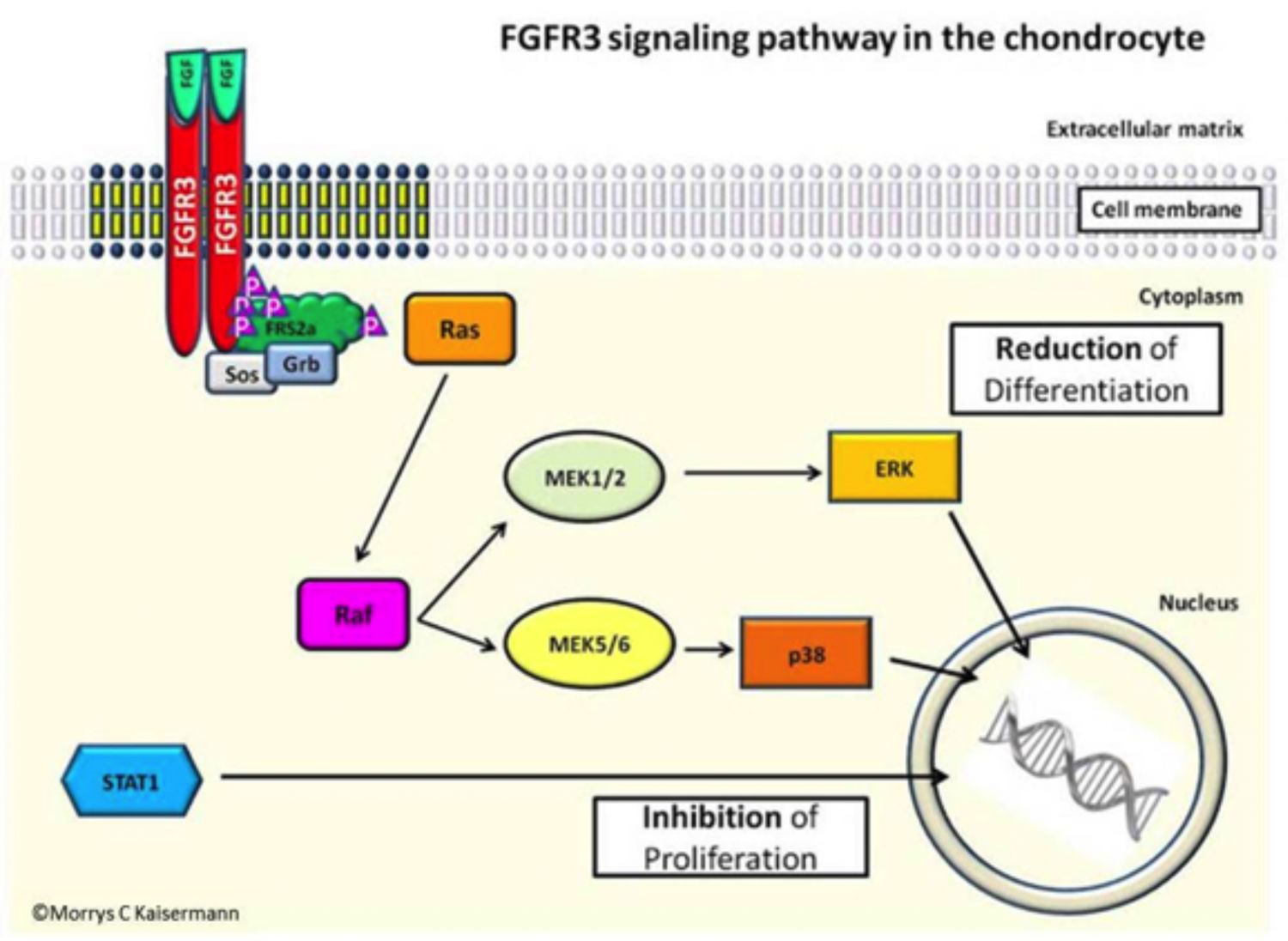
Imagen 3. La vía de señalización del FGFR3 en los condrocitos. Créditos: Tratando la acondroplasia, 2012
En el 80% de los casos la acondroplasia se debe a una mutación espontánea en alguna de las células progenitoras de padres sin antecedentes de mutación, lo que se conoce también como mutación nueva o de novo (Faruki T et al., 2014). Las mutaciones de novo pueden asociarse con edad avanzada de los padres (superior a los 35 años de edad) debido a errores durante la gametogénesis (Natacci F et al., 2008). Para parejas que no tienen mutación y que han tenido un niño con acondroplasia, el riesgo de recurrencia es muy pequeño (<1%) pero no desdeñable. Esto se debe al riesgo de mosaicismo de línea germinal que tiene lugar cuando la mutación solo aparece en el lo aparece en cuando del esperma (Wright M and Irving M, 2011).
La transferencia de la mutación a los descendientes sigue la siguiente pauta: una persona con acondroplasia que planea tener hijos con una pareja que no tiene acondroplasia tiene una probabilidad del 50% de tener un niño con acondroplasia y una probabilidad del 50% de tener (Imagen 4).
Cuando ambos padres tienen acondroplasia la probabilidad de tener un niño sin acondroplasia es del 25%, de tener un niño con acondroplasia es del 50% y de tener un hijo que hereda la mutación genética de ambos padres (llamada acondroplasia severa homocigótica (SADDAN), una condición muy grave que es incompatible con la vida) (Pauli R, 2012), es del 25%.
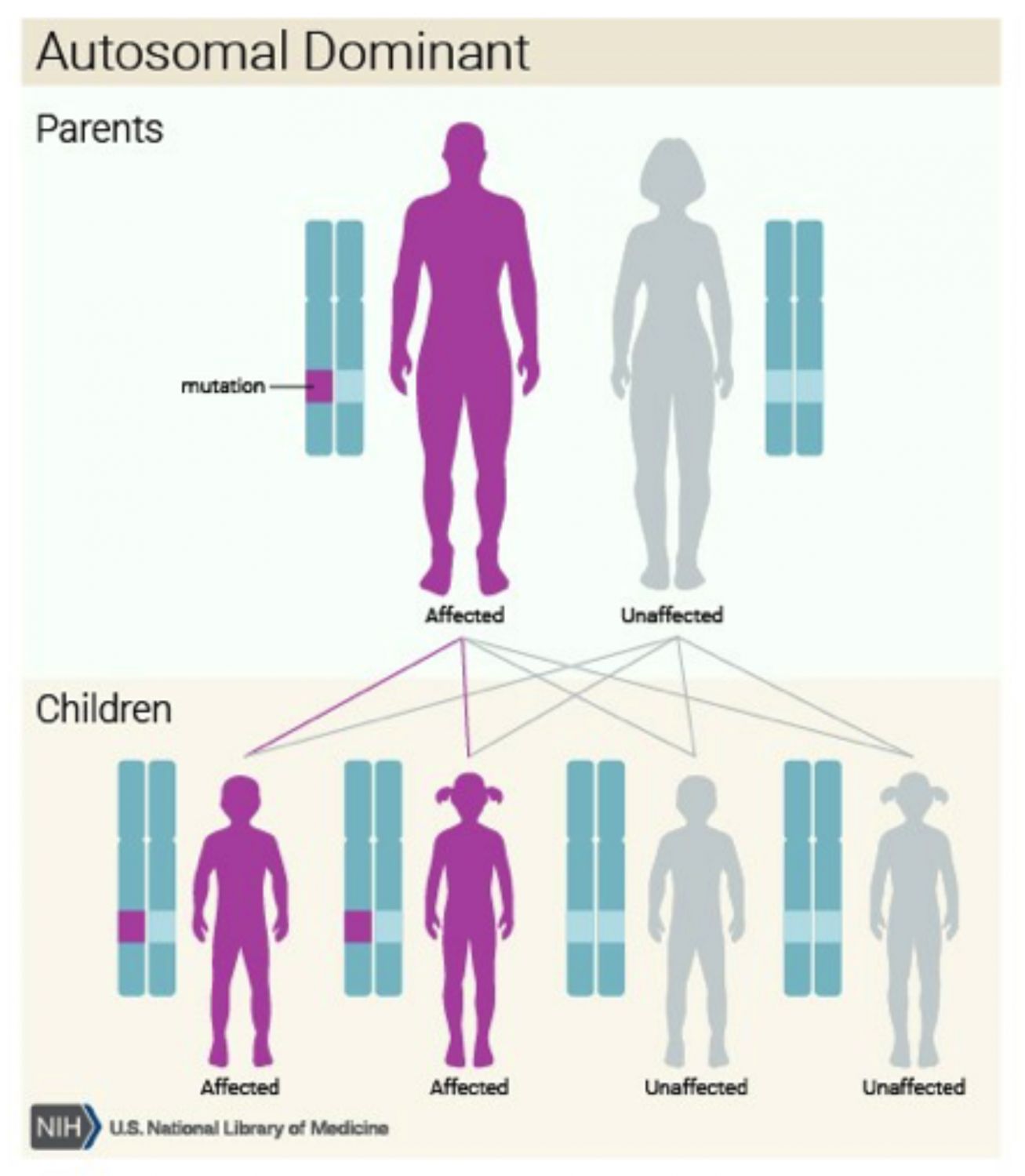
Imagen 4. Esquema de herencia autosómica dominante. Créditos: Genetics Home reference, 2012
BIBLIOGRAFÍA
Faruqi T, Dhawan N, Bahl J, Gupta V, Vohra S, Tu K, Abdelmagid S. Molecular, Phenotypic Aspects and Therapeutic Horizons of Rare Genetic Bone Disorders. BioMed Research International 2014;2014:670842.
He L, Horton W, Hristova K. Physical Basis behind Achondroplasia, the Most Common Form of Human Dwarfism. J Biol Chem 2010;285(39):30103-30114.
Horton W, Hall J, Hecht J. Achondroplasia. Lancet 2007;370: 162-72.
Lee YC, Song IW, Pai YJ, Chen SD, Chen YT. Knock-in human FGFR3 achondroplasia mutation as a mouse model for human skeletal dysplasia. Sci Rep 2017;7:43220.
Narayana J, Horton WA. Molecular Genetics of Achondroplasia. eLS, 2013 DOI: 10.1002/9780470015902.a0024296.
Natacci F, Baffico M, Cavallari U, Bedeschi MF, Mura I, Paffoni A, Setti P, Baldi M, Lalatta F. Clinical Report Germline Mosaicism in Achondroplasia Detected in Sperm DNA of the Father of Three Affected Sibs. Am J Med Genet A 2008;146A:784-786-
Pauli RM. Achondroplasia. 1998 Oct 12 [Updated 2012]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® University of Washington, Seattle; 1993-2017
Wright M, Irving M. Clinical management of achondroplasia. Arch Dis Child, 2012; 97 (2): 129-134.
La Fundación ALPE Acondroplasia fue creada el 24 de enero de 2000 gracias al entusiasmo de varias personas, Carmen Alonso, Miguel López y la familia Press-Lewis, fundamentalmente. La familia Press-Lewis había fundado ProChon Biotech Ltd. en Tel-Aviv (Israel) para la búsqueda de una terapia para la acondroplasia. ProChon fue germen de avances científicos en la investigación de la acondroplasia que dan frutos cada vez más interesantes.
![]()

ENLACES
ACTUALIDAD/EVENTOS/NOTICIAS


CONSULTAS MÉDICAS
Fundación ALPE Acondroplasia
Calle Conde Real Agrado, 2
33205 Gijón